
Imprintingul genetic (genomic) ?i disomia uniparental?
Fiecare individ mo?tene?te 2 copii ale fiec?rei gene – una de la mam? ?i una de la tat?.
În mod normal, ambele copii ale fiecărei gene sunt active, însă în unele cazuri, doar una dintre copii este activă. Care dintre copii este activă depinde de originea maternă sau paternă: unele gene sunt active doar dacă sunt moștenite de la tată, altele dacă sunt moștenite de la mamă. Acest fenomen se numește imprinting genetic (imprinting genomic) și constă în exprimarea diferențiată a genelor, în funcția de originea lor parentală.
În genele amprentate genomic, originea parenterală a lor este etichetată în procesul de formare a spermatozoizilor și ovulelor, prin metilare. Prezența grupărilor metil în anumite segmente ale ADN-ului indică dacă gena a fost moștenită de la mamă sau de la tată. Adăugarea sau îndepărtarea grupărilor metil poate fi folosită și pentru a controla activitatea genelor. Doar un mic procent dintre toate genele umane suferă procesul de imprinting genetic. Cercetătorii nu au descoperit încă de ce unele gene sunt amprentate și altele nu, însă știu faptul că aceste gene au tendința de a se aglomera în aceleași regiuni ale cromozomilor. Au fost identificare 2 regiuni majore de acumulare de gene amprentate – una pe brațul scurt (p) al cromozomului 11 (la poziția 11p15) și una pe brațul lung (q) al cromozomului 15 (în regiunea 15q11-15q13).
Disomia uniparentală apare atunci când un individ moștenește 2 copii ale unui cromozom sau porțiuni ale unui cromozom, doar de la unul dintre părinți, și nicio copie de la celălalt părinte. Acest fenomen poate apărea fie ca un eveniment aleator în procesul de formare a spermatozoizilor sau ovulelor, fie precoce în timpul dezvoltării fetale. În multe cazuri, disomia uniparentală nu are niciun efect asupra sănătății sau dezvoltării individului. Deoarece majoritatea genelor nu sunt amprentate, nu contează dacă o persoană moștenește ambele copii de la un părinte în loc de câte o copie de la fiecare părinte. În unele cazuri însă, există o diferență dacă o genă este moștenită de la mamă sau de la tată. Unui individ cu disomie uniparentală îi poate lipsi copia activă a unei gene amprentată genomic, ceea ce poate duce la retard în dezvoltare psihomotorie sau alte probleme medicale.
Există mai multe afecțiuni genetice apărute ca și consecință a disomiei uniparentale sau perturbării unui imprinting genetic normal: sindromul Prader-Willi (caracterizat prin apetit necontrolat și obezitate) și sindromul Angelman (caracterizat prin retard mental și tulburări de vorbire) – ambele implicând gene de pe brațul lung al cromozomuluin 15, și sindromul Beckwith-Wiedemann (caracterizat prin creștere accelerată și risc crescut de tumori) – asociat cu anomalii de amprentare genomică a genelor de pe brațul scurt al cromozomului 11.
Sindromul Prader-Willi
Sindromul Prader-Willi este o anomalie cromozomială secundară deleției unor gene de pe brațul lung al cromozomului 15 de origine paternă sau disomiei materne a cromozomuluin 15 (în care ambii cromozomi 15 sunt de origine maternă). Sindromul a fost descris pentru prima oară în 1956, însă modificările genetice asociate au fost descoperite de cercetători abia prin anii 1980. Dr. David Ledbetter a fost cel care în 1981 a pus în evidență faptul că o mare parte dintre pacienții cu sindrom Prader-Willi prezentau o deleție a aceluiași segment de gene de pe cromozomul 15. Frecvența acestei boli genetice este de 1:15.000 nou-născuți, afectează în mod egal ambele sexe și a fost identificată la toate rasele. Principalele trăsături caracteristice sindromului Prader-Willi sunt: hipotonia, statura mică, polifagia, obezitatea, hipogonadismul, strabismul, retardul mental ușor, mâinile și picioarele mici. La nivel mondial, există peste 400.000 persoane care au sindrom Prader-Willi.
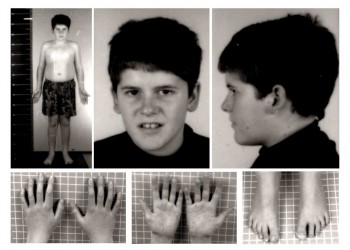
Fenotipul sindromului Prader-Willi
Caracteristicile clinice ale pacienților cu sindrom Prader-Willi depind de vârstă:
- in utero: mișcări fetale reduse, poziție fetală anormală, ocazional polihidramnios (exces de lichid aminiotic)
- la naștere: naștere prin cezariană, letargie, hipotonie, dificultăți de alimentație (tonusul muscular scăzut afectează reflexul de supt), dificultăți de respirație, hipogonadism
- sugari: falimentul creșterii (secundar dificultăților de alimentație), retard în creștere și dezvoltare intelectuală, somn excesiv, strabism, scolioză (uzual nedetectată la naștere).
- copilărie: întârziere în vorbire, coordonare fizică deficitară, hiperfagie de la vârsta de 2-4 ani (opus față de dificultățile de alimentație din perioada de sugar), creștere ponderală excesivă, tulburări de somn, scolioză.
- adolescență: pubertate întârziată, statură mică, obezitate, flexibilitate extremă.
- adult: infertilitate (la ambele sexe), hipogonadism, păr pubian rar, obezitate, hipotonie, dificultăți de învățare, dezvoltare intelectuală la limită (există și cazuri de inteligență medie), predispoziție la diabet zaharat, flexibilitate extremă.
- aspect fizic general la adulți: rădăcina nasului proeminentă, mâini și picioare mici, cu degete ascuțite, piele subțire (care se rănește ușor), țesut adipos în exces (obezitate predominant centrală), frunte îngustă și înaltă, tegumente și pilozitate de culoare deschisă (comparativ cu ceilalți membri ai familiei), ciupirea pielii (skin picking), vergeturi, dezvoltare motorie întârziată, lipsă a dezvoltării sexuale complete, ochi ca migdala, comisuri bucale oblice inferior.
Diagnostic
Diagnosticul de sindrom Prader-Willi se pune pe baza elementelor clinice și se confirmă prin testare genetică; testarea este recomandată pentru nou-născuții cu hipotonie marcată. Diagnosticul precoce al sindromului Prader-Willi permite intervenția medicală timpurie și prescrierea de hormon de creștere (se recomandă injecții zilnice cu hormon de creștere – GH recombinant). Tratamentul cu hormon de creștere sigură o dezvoltare normală și o creștere a masei musculare, putând totodată diminua preocuparea pentru alimentație și creșterea ponderală excesivă.
Principala metodă diagnostică este testul de metilare a ADN, care detectează absența paternală a regiunii pentru sindromul Prader-Willi/Angelmann la nivelul cromozomului 15q11-q13. Acuratețea testului este de peste 97%. Diagnosticul genetic este important îndeosebi pentru indivizii a căror vârstă este prea mică pentru a manifesta suficiente caracteristici pentru un diagnostic clinic și pentru cei cu manifestări atipice. Formele atipice de sindrom Prader-Willi pot apărea datorită unui traumatism la naștere, la trecerea prin canalul vaginal.
Pe baza elementelor clinice este necesar de făcut diagnosticul diferențial cu sindromul Down, cu care este frecvent confundat datorită incidenței crescute a acestuia comparativ cu sindromul Prader-Willi. Obezitatea marcată poate apărea și în sindromul Down datorită unor tulburări comportamentale.
Diagnosticul clinic se face pe baza unui scor care cuprinde evaluare unor criterii majore și minore (1 criteriu major=1 punct, 2 criterii minore=1 punct). Un scor de minim 5 puncte (dintre care 4 obținute prin criterii majore) până la vârsta de 3 ani sau de minim 8 puncte (dintre care 5 obținute prin criterii majore) după vârsta de 3 ani pun diagnosticul de sindrom Prader-Willi.
i) Criterii majore:
- hipotonie neonatală și infantilă, supt dificil
- dificultăți de alimentare și greutate mică
- creștere ponderală rapidă și excesivă în perioada 1-6 ani
- hiperfagie
- facies caracteristic (îngustarea diametrului bifrontal, ochi migdalați, gură mică cu buză superioară subțire, comisuri bucale coborâte)
- hipogonadism, dependent de vârstă
- retard mental ușor-moderat.
ii) Criterii minore:
- scăderea intensității mișcărilor fetale, letargie infantilă sau plânset slab corectate cu vârsta
- tulburări comportamentale: accese de furie, reacții violente, atitudine obsesivă, tendințe spre argumentare, opoziție, rigiditate, posesivitate, încăpățânare, mitomanie, furt
- tulburări de somn, apnee în timpul somnului
- statură mică, hipopigmentarea părului și pielii
- mâini și picioare mici, mâini înguste în continuarea liniară a cubitusului
- anomalii oculare: ezotropie (strabism convergent), miopie
- salivă vâscoasă, cruste ale comisurilor bucale
- defecte de articulare a cuvintelor, ciupirea pielii
Diagnosticul pe baza scorului clinic trebuie să fie urmat de cariotipare pentru identificarea deleției sau rearanjamentelor cromozomiale de la nivelul 15q11-q13. Dacă studiile genetice nu evidențiază modificări cromozomiale, se indică efectuarea testelor moleculare (test FISH, test de metilare a ADN).
Aspecte genetice
În sindromul Prader-Willi regiunea q11-q13 de la nivelul cromozomului 15 prezintă o anomalie: mai multe gene care trebuie să provină de la tatăl copilului pentru a funcționa normal, fie lipsesc, fie sunt inactivate în cromozomul patern. O situație similară se întâlnește în cazul în care la nivelul aceleiași regiuni există o genă amprentată pentru a funcționa numai la nivelul cromozomului matern; dacă ea lipsește sau este inactivată la nivelul cromozomului matern, apare sindromul Angelman. Așadar, pierdere materialului genetic de origine paternă, amprentat, situat pe brațul lung al cromozomului 15 în regiunea 11q-13q determină sindromul Prader-Willi, iar pierderea materialului genetic din aceeași regiune a cromozomului 15 matern determină sindromul Angelman. Deoarece anomaliile genetice sunt localizate în același segment ale brațului lung al cromozomului 15, cele 2 sindroame sunt denumite “sindroame surori”. Sindroamele Angelman și Prader-Willi sunt primele afecțiuni genetice demonstrate la om a fi asociate cu defecte de imprinting genetic.
Există mai multe anomalii genetice care pot determina sindrom Prader-Willi:
- deleție a regiunii 15q11-q13 de pe cromozomul patern – 70% din cazuri
- disomie uniparentală (ambii cromozomi 15 de origine maternă) – 25% din cazuri
- defecte ale centrului de amprentare – 5% din cazuri
- rearanjamente cromozomiale (translocații), mutații sporadice – sub 1% din cazuri
Cercetările actuale sunt îndreptate către descoperirea genelor specifice implicate în sindromul Prader-Willi și care este funcția acestor gene.
Sfatul genetic
Majoritatea cazurilor de sindrom Prader-Willi sunt sporadice, părinții fiind complet sănătoși. Riscul unui cuplu cu un copil Prader-Willi de a avea un nou copil afectat depinde de mecanismul genetic cauzal. În cazul în mecanismul este deleție a regiunii critice 15q11-15q13 sau disomie uniparentală maternă, riscul de a avea încă un copil cu sindrom Prader-Willi este sub 1%. Dacă mecanismul este o mutație a centrului de amprentare riscul este de până la 50%, iar dacă se identifică o translocație parentală (prezentă și la copilul afectat) riscul este de până la 25%. Există posibilitatea testării prenatale pentru toate mecanismele genetice incriminate. Deși majoritatea pacienților cu sindrom Prader-Willi nu se reproduc, riscul de a avea un copil cu sindrom Prader-Willi (dacă tatăl este afectat) sau Angelman (dacă mama este afectată) este de 50%.
Teste genetice
Testele genetice indicate în cazul unei suspiciuni de sindrom Prader-Willi sunt:
- analiză cromozomială cu rezoluție înaltă (examinare la microscop): determină regiuni extinse de deleții, translocații sau anomalii numerice cromozomiale (cromozomi supranumerari). Nu poate evidenția deleții mici, disomia uniparentală sau mutațiile de inactivare.
- FISH (fluorescence in situ hybridization): este un test molecular care poate evidenția orice deleție cromozomială. Dezavantajul metodei este că nu identifică disomia uniparentală (UPD), mutațiile de inactivare și nici nu poate preciza părintele de la care provine mutația (astfel, o deleție la nivelul cromozomului 15 poate însemna fie sindrom Prader-Willi, fie sindrom Angelman). Se poate practica simultan cu analiza cromozomială.
- studii de polimorfism ADN: se efectuează pentru detectarea disomiei uniparentale și presupune recoltarea de sânge atât de la copil, cât și de la ambii părinți. Poate preciza de la care părinte provine cromozomul 15 (dacă ambii cromozomi sunt de la tată este vorba despre sindromul Angelman, dacă ambii cromozomi sunt de la mamă este vorba despre sindromul Prader-Willi). Nu poate evidenția mutațiile de inactivare sau anumite deleții.
- testul de metilare a ADN: are o acuratețe de peste 99% în confirmarea sau excluderea sindromului Prader-Willi. Evidențiază modelul de inactivare a regiunii PW/A 15q11-q13, rezultatul putând indica fie un model monștenit numai de la mamă – deleție, disomie uniparentală sau mutație de inactivare (când este vorba despre sindrom Prader-Willi), fie un model moștenit numai de la tată (când este vorba despre sindrom Angelman). Rezultatele normale indică modele moștenite de la ambii părinți.
Testarea genetică este recomandată în următoarele situații:
- sugar cu tonus muscular scăzut (supt slab) și dacă este băiat, cu testiculi necoborâți
- copil sau adult cu elemente clinice sugestive pentru sindrom Prader-Willi, conform scorului diagnostic
După confirmarea genetică a sindromului Prader-Willi, se recomandă continuarea investigațiilor pentru stabilirea exactă a mecanismului cauzal.
În cazul unei suspiciuni clinice de sindrom Prader-Willi, se recomandă inițial un test FISH pentru depistarea unei deleții la nivelul cromozomului 15 – dacă acesta este pozitiv se confirmă diagnosticul, dacă este negativ – se efectuează un test de metilare ADN. În cazul în care după un FISH negativ, testul de metilare ADN confirmă sindromul, sunt necesare investigații suplimentare pentru a detecta mecanismul cauzal – mutație de inactivare (imprinting) sau disomie uniparentală. În situația în care scorul de diagnostic clinic este insuficient, dar există elemente sugestive pentru sindrom Prader-Willi, se poate începe direct cu testul de metilare ADN - dacă este pozitiv se vor continua investigațiile pentru determinarea mecanismului genetic, dacă este negativ se exclude cu probabilitate de 99% diagnosticul.
Diagnostic prenatal
Diagnosticul prenatal este recomandat familiilor în care există deja un copil cu sindrom Prader-Willi, după acordarea sfatului genetic cu privire la riscul de recurență (crescut în cazul în mecanismul cauzal l copilul afectat este un defect de amprentare sau o translocație echilibrată cu punct de ruptură în regiunea critică). O altă indicație de testare prenatală pentru sindromul Prader-Willi este atunci când apar anomalii cromozomiale la amniocenteză sau biopsia de vilozități coriale (ex. când BVP arată un mozaic de trisomie 15 – anumite celule fetale prezintă 3 cromozomi 15, trebuie efectuat un test molecular pentru disomia uniparentală maternă care poate arăta dacă fătul va avea sindrom Prader-Willi). Testarea este recomandată și familiilor care sunt îngrijorate de riscul individual de a avea un copil cu sindrom Prader-Willi, după acordarea în prealabil a sfatului genetic.
Evoluție. Tratament și îngrijire
Pacienții cu sindrom Prader-Willi ajung la vârsta adultă, însă datorită deficitelor neuro-cognitive și comportamentale ei necesită îngrijiri speciale. Statistic, performanța intelectuală se încadrează astfel (Curfs și Frym, 1992): 5% - IQ peste 85 (inteligență medie-submedie), 27% - IQ între 70 și 85 (performanță intelectuală la limită), 34% - IQ între 50 și 70 (dizabilitate intelectuală ușoară), 27% - IQ între 35 și 50 (dizabilitate intelectuală moderată), 5% - IQ între 20 și 35 (dizabilitate intelectuală severă), 1% - IQ sub 20 (dizabilitate intelectuală profundă). Un alt studiu (Cassidy) a relevat că 40% dintre indivizii cu sindrom Prader-Willi au inteligență submedie sau la limită. În grupuri de lucru de dimensiuni mici, individualizate, acești pacienți pot avea un randament social bun.
În ceea ce privește profilul cognitiv, pacienții cu sindrom Prader-Willi prezintă un important deficit de vorbire (uneori afectată și de hipernazalitate), al abilității de scriere și efectuării de operații aritmetice, memoriei vizuale și auditive pe termen scurt și capacității de concentrare. Din punct de vedere comportamental, pacienții prezintă un apetit extrem și insațiabil, ceea ce duce adesea la obezitate morbidă. Actualmente nu există un consens în ceea ce privește cauza acestui simptom particular, explicația fiind oferită de faptul că anomaliile genetice de la nivelul cromozomului 15 interferă cu funcționarea normală a hipotalamusului (care printre altele, coordonează și apetitul); totuși, investigarea post-mortem nu a relevat niciun defect organic al hipotalamusului. Totodată, pacienții cu sindrom Prader-Willi prezintă niveluri crescute de grelină, care contribuie la apetitul crescut și hiperfagie.
Din punct de vedere endocrin, indivizii prezintă un deficit de hormon de creștere susținut de: statura mică, obezitate, masă nelipidică redusă (fat free mass, FFM), densitate minerală scăzută. Persoanele cu Prader-Willi mai prezintă hipogonadism (testicule necoborâte în scrot la băieți, adrenarhă prematură la sexul feminin). Criptorhidia se poate rezolva spontan sau necesită intervenție terapeutică hormonală sau chirurgicală. Adrenarha necesită tratament de substituție hormonală.
Tratamentul este unul suportiv, constând în măsuri menite să amelioreze simptomatologia și să permită o dezvoltare cât mai aproape de normal:
- în perioada de sugar se vor folosi tehnici speciale de alăptat (inclusiv gavaj)
- folosirea de tehnici de îmbunătățire a tonusului muscular la copilul mic
- mediu de învățare adecvat în perioada de școlar
- administrarea de hormon de creștere în copilărie asigură o dezvoltare staturală și ponderală normale și o masă musculară adecvată
- dietă hipocalorică, program de exerciții fizice, limitarea accesului la alimente (măsuri destinate restricționării hiperfagiei care duce la obezitate)
- monitorizarea apariției scoliozei
- antipsihotice (inhibitori de serotonină) pentru tulburările comportamentale
- suplimente de calciu pentru prevenirea osteoporozei
- monitorizarea afecțiunilor acute al căror diagnostic este temporizat datorită insensibilității la durere a pacienților
- limitarea efectelor nocive ale obezității: apnee obstructivă în somn, boli cardiovasculare, diabet zaharat tip II, insuficiență venoasă cronică a membrelor inferioare
Comments